
diff options
Diffstat (limited to 'general')
| -rw-r--r-- | general/brand/aging/home.md | 15 | ||||
| -rw-r--r-- | general/brand/aging/um-het3.png | bin | 0 -> 880629 bytes | |||
| -rw-r--r-- | general/brand/gnqa/gnqa.md | 20 | ||||
| -rw-r--r-- | general/brand/gnqa/imgs/integration.png | bin | 0 -> 247838 bytes | |||
| -rw-r--r-- | general/brand/gnqa/imgs/pubmed-ref.png | bin | 0 -> 109781 bytes | |||
| -rw-r--r-- | general/brand/gnqa/imgs/refs.png | bin | 0 -> 95656 bytes | |||
| -rw-r--r-- | general/brand/gnqa/imgs/workflow.png | bin | 0 -> 97521 bytes | |||
| -rw-r--r-- | general/glossary/glossary.md | 116 | ||||
| -rw-r--r-- | general/help/facilities.md | 80 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
9 files changed, 116 insertions, 115 deletions
diff --git a/general/brand/aging/home.md b/general/brand/aging/home.md index 33c18e1..17ca578 100644 --- a/general/brand/aging/home.md +++ b/general/brand/aging/home.md @@ -1,5 +1,14 @@ -New aging portal! This is initial work for providing a full community webservice for aging. +New UMHET3 and aging portal! This is initial work for providing a full community webservice for aging. +- [https://doi.org/10.1101/2025.04.27.649857](https://doi.org/10.1101/2025.04.27.649857). +**Genetic Modulation of Lifespan: Dynamic Effects, Sex Differences, and Body Weight Trade-offs** -This is a stub until David edits it! Or Rob. See this link to [edit](https://github.com/genenetwork/gn-docs/edit/master/general/brand/aging/home.md). +> **Abstract**: The dynamics of lifespan are shaped by DNA variants that exert effects at different ages. We have mapped genetic loci that modulate age-specific mortality using an actuarial approach. We started with an initial population of 6,438 pubescent siblings and ended with a survivorship of 559 mice that lived to at least 1100 days. Twenty-nine Vita loci dynamically modulate the mean lifespan of survivorships with strong age- and sex-specific effects. Fourteen have relatively steady effects on mortality while other loci act forcefully only early or late in life and with polarities of effects that invert. A distinct set of 19 Soma loci shape the negative correlation between weights of young adults with their life expectancies—much more strongly so in males than females. Another set of 11 Soma loci shape the positive correlation between weights at older ages with life expectancies. The Vita and Soma loci share 289 age-dependent epistatic interactions (LODs ≥3.8) but fewer than 4% are common to both sexes. [More...](https://doi.org/10.1101/2025.04.27.649857). -For markdown check [the docs](https://commonmark.org/help/). To embed images you can use a link somewhere from the internet or check it into the gn-doc repo and I'll add a link. +We provide two examples of how to move from maps toward potential mechanisms. Our findings provide an empirical bridge between evolutionary theories on aging and genetic and molecular causes. These loci and their interactions are key to begin to understand the impact of interventions that may extend healthy lifespan in mice and even in humans. + + + +- Data files are available [here](https://files.genenetwork.org/current/umhet3_2025/). +- Code is available at [https://github.com/DannyArends/UM-HET3/](https://github.com/DannyArends/UM-HET3/) + +See also GeneNetwork.org [examples on aging](https://aging.genenetwork.org/). diff --git a/general/brand/aging/um-het3.png b/general/brand/aging/um-het3.png new file mode 100644 index 0000000..5c762ce --- /dev/null +++ b/general/brand/aging/um-het3.png Binary files differdiff --git a/general/brand/gnqa/gnqa.md b/general/brand/gnqa/gnqa.md new file mode 100644 index 0000000..bdd559c --- /dev/null +++ b/general/brand/gnqa/gnqa.md @@ -0,0 +1,20 @@ +# GNQA the Genenetwork.org Question Answer System + +## Purpose + +Genenetwork.org (GN) is a site housing tens of terabytes of experimental genetic and genomic information on many species, most notably mouse and human. In its constantly evolving state the masterminds behind this systems genetics service look to make it easier to use for non-experts or citizen scientists. To this end GNQA, a retrieval augmented generation service, is under development to support knowledge communication and research understanding. + +### Accessing GNQA +In order to use GNQA you must be a registered user of genenetwork.org. If you have not yet registered, do so [here](https://genenetwork.org/oauth2/user/register). +The GN registration page with have you verify the email address you used for registration. Once your email has been confirmed you can log in to genenetwork.org. +There are two ways to query GNQA: the 1st is by using the genenetwork.org global search, the 2nd is by visiting [GNQA directly](https://genenetwork.org/gnqna). +When using the global search, GNQA results are presented in a small panel above the table of records. +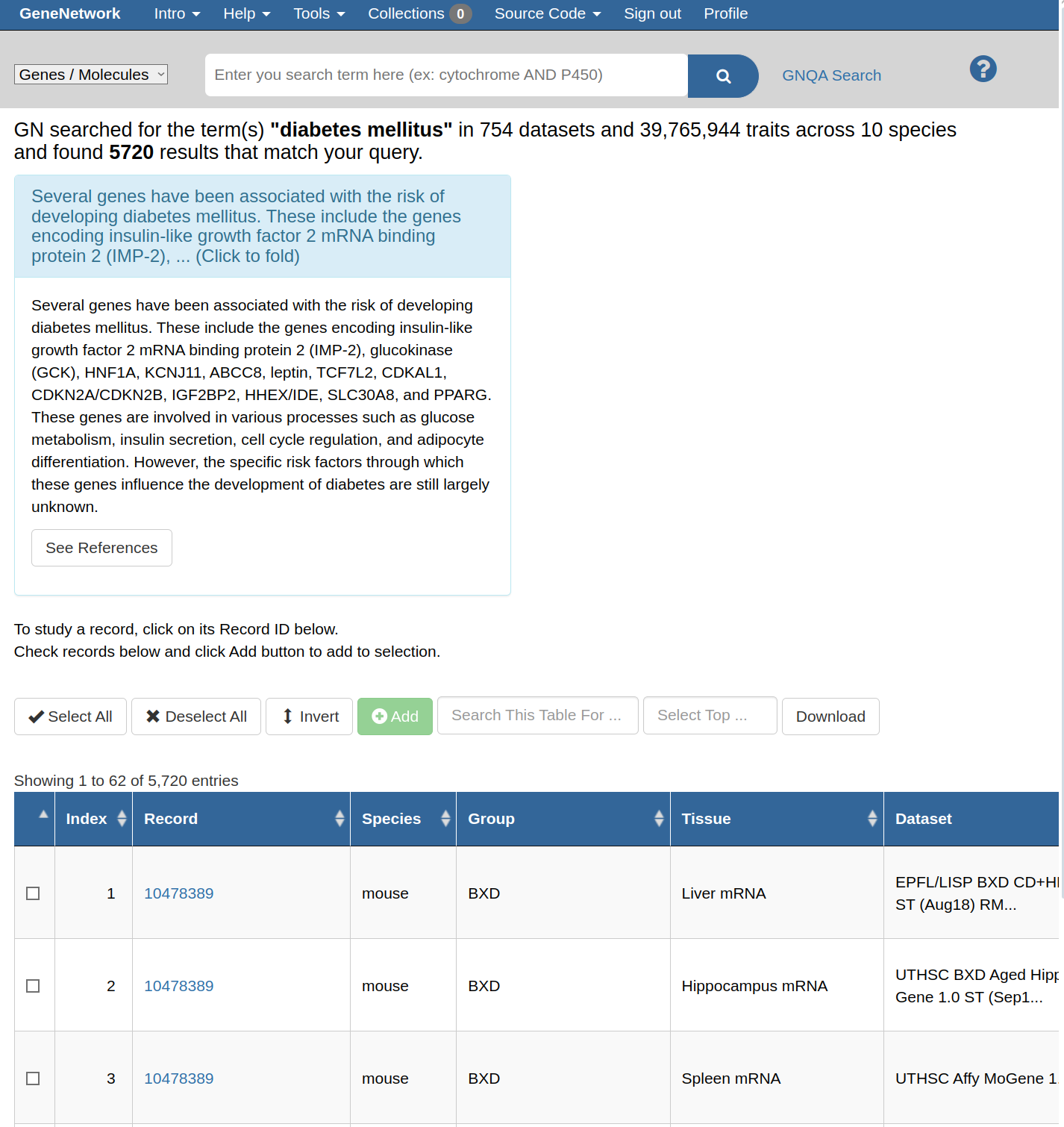 + + +## Knowledge Communication & Research Understanding + +GNQA is a tool utilizing a large language model and a separately maintained scientific knowledge base with specially and specifically curated data. The curated data is currently focused on three main areas: GN research, genomics of aging, and the genomics of diabetes. We chose each area based on our research interests. GNQA as it currently exists, as of 16 January 2025, is comprised of 3000 research tomes in our listed areas of interest. The GN global search bar returns a table of often several thousand results and links to datasets related to ones query, whereas GNQA queries its knowledge base of research documents and books to return an answer to the query along with a list of references, real... not hallucinated. Some references are presented with links to the research documents on [PubMed](https://www.ncbi.nlm.nih.gov/pubmed "The destination for science research"). + +### System Workflow +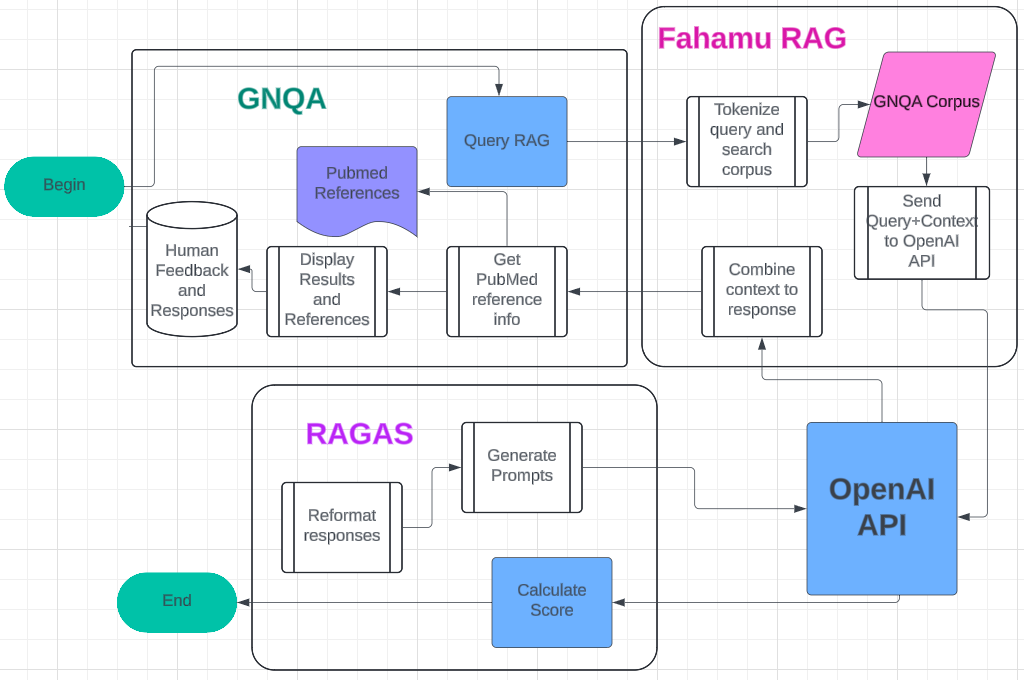 diff --git a/general/brand/gnqa/imgs/integration.png b/general/brand/gnqa/imgs/integration.png new file mode 100644 index 0000000..d08039f --- /dev/null +++ b/general/brand/gnqa/imgs/integration.png Binary files differdiff --git a/general/brand/gnqa/imgs/pubmed-ref.png b/general/brand/gnqa/imgs/pubmed-ref.png new file mode 100644 index 0000000..8e72933 --- /dev/null +++ b/general/brand/gnqa/imgs/pubmed-ref.png Binary files differdiff --git a/general/brand/gnqa/imgs/refs.png b/general/brand/gnqa/imgs/refs.png new file mode 100644 index 0000000..819da62 --- /dev/null +++ b/general/brand/gnqa/imgs/refs.png Binary files differdiff --git a/general/brand/gnqa/imgs/workflow.png b/general/brand/gnqa/imgs/workflow.png new file mode 100644 index 0000000..fdc9916 --- /dev/null +++ b/general/brand/gnqa/imgs/workflow.png Binary files differdiff --git a/general/glossary/glossary.md b/general/glossary/glossary.md index 883e338..1d66a21 100644 --- a/general/glossary/glossary.md +++ b/general/glossary/glossary.md @@ -43,42 +43,42 @@ KNOWN PROBLEMS and INTERPRETATION of BOOTSTRAP RESULTS: The reliability of boots There is a known flaw in the way in which GeneNetwork displays bootstrap results (Sept 2011). If a map has two or more adjacent markers with identical LOD score and identical strain distribution patterns, all of the bootstrap results are assigned incorrectly to just one of the "twin" markers. This results in a false perception of precision. -QTL mapping methods can be highly sensitive to cases with very high or very low phenotype values (outliers). The bootstrap method does not provide protection against the effects of outliers and their effects on QTL maps. Make sure you review your data for outliers before mapping. Options include (1) Do nothing, (2) Delete the outliers and see what happens to your maps, (3) [Winsorize](http://en.wikipedia.org/wiki/Winsorising) the values of the outliers. You might try all three options and determine if your main results are stable or not. With small samples or extreme outliers, you may find the mapping results to be quite volatile. In general, if the results (QTL position or value) depend highly on one or two outliers (5-10% of the samples) then you should probably delete or winsorize the outliers. [Williams RW, Oct 15, 2004, Mar 15, 2008, Mar 26, 2008; Sept 2011] - +QTL mapping methods can be highly sensitive to cases with very high or very low phenotype values (outliers). The bootstrap method does not provide protection against the effects of outliers and their effects on QTL maps. Make sure you review your data for outliers before mapping. Options include (1) Do nothing, (2) Delete the outliers and see what happens to your maps, (3) [Winsorize](http://en.wikipedia.org/wiki/Winsorising) the values of the outliers. You might try all three options and determine if your main results are stable or not. With small samples or extreme outliers, you may find the mapping results to be quite volatile. In general, if the results (QTL position or value) depend highly on one or two outliers (5-10% of the samples) then you should probably delete or winsorize the outliers. [Williams RW, Oct 15, 2004, Mar 15, 2008, Mar 26, 2008; Sept 2011] + [Go back to index](#index) <div id="c"></div> #### CEL and DAT Files (Affymetrix): -Array data begin as raw image files that are generated using a confocal microscope and video system. Affymetrix refers to these image data files as DAT files. The DAT image needs to be registered to a template that assigns pixel values to expected array coordinates (cells). The result is an assignment of a set of image intensity values (pixel intensities) to each probe. For example, each cell/probe value generated using Affymetrix arrays is associated with approximately 36 pixels (a 6x6 set of pixels, usually with an effective 11 or 12-bit range of intensity). Affymetrix uses a method that simply ranks the values of these pixels and picks as the "representative value" the pixel that is has rank 24 from low to high. The range of variation in intensity amoung these ranked pixels provides a way to estimate the error of the estimate. The Affymetrix CEL files therefore consist of XY coordinates, the consensus value, and an error term. [Williams RW, April 30, 2005] +Array data begin as raw image files that are generated using a confocal microscope and video system. Affymetrix refers to these image data files as DAT files. The DAT image needs to be registered to a template that assigns pixel values to expected array coordinates (cells). The result is an assignment of a set of image intensity values (pixel intensities) to each probe. For example, each cell/probe value generated using Affymetrix arrays is associated with approximately 36 pixels (a 6x6 set of pixels, usually with an effective 11 or 12-bit range of intensity). Affymetrix uses a method that simply ranks the values of these pixels and picks as the "representative value" the pixel that is has rank 24 from low to high. The range of variation in intensity amoung these ranked pixels provides a way to estimate the error of the estimate. The Affymetrix CEL files therefore consist of XY coordinates, the consensus value, and an error term. [Williams RW, April 30, 2005] #### Cluster Map or QTL Cluster Map: -Cluster maps are sets of QTL maps for a group of traits. The QTL maps for the individual traits (up to 100) are run side by side to enable easy detection of common and unique QTLs. Traits are clustered along one axis of the map by phenotypic similarity (hierarchical clustering) using the Pearson product-moment correlation r as a measurement of similarity (we plot 1-r as the distance). Traits that are positively correlated will be located near to each other. The genome location is shown along the other, long axis of the cluster map, marker by marker, from Chromosome 1 to Chromosome X. Colors are used to encode the probability of linkage, as well as the additive effect polarity of alleles at each marker. These QTL maps are computed using the fast Marker Regression algorithm. P values for each trait are computed by permuting each trait 1000 times. Cluster maps could be considered trait gels because each lane is loaded with a trait that is run out along the genome. Cluster maps are a unique feature of the GeneNetwork developed by Elissa Chesler and implemented in WebQTL by J Wang and RW Williams, April 2004. [Williams RW, Dec 23, 2004, rev June 15, 2006 RWW]. +Cluster maps are sets of QTL maps for a group of traits. The QTL maps for the individual traits (up to 100) are run side by side to enable easy detection of common and unique QTLs. Traits are clustered along one axis of the map by phenotypic similarity (hierarchical clustering) using the Pearson product-moment correlation r as a measurement of similarity (we plot 1-r as the distance). Traits that are positively correlated will be located near to each other. The genome location is shown along the other, long axis of the cluster map, marker by marker, from Chromosome 1 to Chromosome X. Colors are used to encode the probability of linkage, as well as the additive effect polarity of alleles at each marker. These QTL maps are computed using the fast Marker Regression algorithm. P values for each trait are computed by permuting each trait 1000 times. Cluster maps could be considered trait gels because each lane is loaded with a trait that is run out along the genome. Cluster maps are a unique feature of the GeneNetwork developed by Elissa Chesler and implemented in WebQTL by J Wang and RW Williams, April 2004. [Williams RW, Dec 23, 2004, rev June 15, 2006 RWW]. #### Collections and Trait Collections: -One of the most powerful features of GeneNetwork (GN) is the ability to study large sets of traits that have been measured using a common genetic reference population or panel (GRP). This is one of the key requirements of systems genetics--many traits studied in common. Under the main GN menu **Search** heading you will see a link to **Trait Collections**. You can assemble you own collection for any GRP by simply adding items using the Add to Collection button that you will find in many windows. Once you have a collection you will have access to a new set of tools for analysis of your collection, including **QTL Cluster Map, Network Graph, Correlation Matrix**, and **Compare Correlates**. [Williams RW, April 7, 2006] +One of the most powerful features of GeneNetwork (GN) is the ability to study large sets of traits that have been measured using a common genetic reference population or panel (GRP). This is one of the key requirements of systems genetics--many traits studied in common. Under the main GN menu **Search** heading you will see a link to **Trait Collections**. You can assemble you own collection for any GRP by simply adding items using the Add to Collection button that you will find in many windows. Once you have a collection you will have access to a new set of tools for analysis of your collection, including **QTL Cluster Map, Network Graph, Correlation Matrix**, and **Compare Correlates**. [Williams RW, April 7, 2006] #### Complex Trait Analysis: -Complex trait analysis is the study of multiple causes of variation of phenotypes within species. Essentially all traits that vary within a population are modulated by a set of genetic and environmental factors. Finding and characterizing the multiple genetic sources of variation is referred to as "genetic dissection" or "QTL mapping." In comparison, complex trait analysis has a slightly broader focus and includes the analysis of the effects of environmental perturbation, and gene-by-environment interactions on phenotypes; the "norm of reaction." Please also see the glossary term "Systems Genetics." [Williams RW, April 12, 2005] +Complex trait analysis is the study of multiple causes of variation of phenotypes within species. Essentially all traits that vary within a population are modulated by a set of genetic and environmental factors. Finding and characterizing the multiple genetic sources of variation is referred to as "genetic dissection" or "QTL mapping." In comparison, complex trait analysis has a slightly broader focus and includes the analysis of the effects of environmental perturbation, and gene-by-environment interactions on phenotypes; the "norm of reaction." Please also see the glossary term "Systems Genetics." [Williams RW, April 12, 2005] #### Composite Interval Mapping: -Composite interval mapping is a method of mapping chromosomal regions that controls for some fraction of the genetic variability in a quantitative trait. Unlike simple interval mapping, composite interval mapping usually controls for variation produced at one or more background marker loci. These background markers are generally chosen because they are already known to be close to the location of a significant QTL. By factoring out a portion of the genetic variance produced by a major QTL, one can occasionally detect secondary QTLs. WebQTL allows users to control for a single background marker. To select this marker, first run the **Marker Regression** analysis (and if necessary, check the box labeled display all LRS, select the appropriate locus, and the click on either **Composite Interval Mapping** or **Composite Regression**. A more powerful and effective alternative to composite interval mapping is pair-scan analysis. This latter method takes into accounts (models) both the independent effects of two loci and possible two-locus epistatic interactions. [Williams RW, Dec 20, 2004] +Composite interval mapping is a method of mapping chromosomal regions that controls for some fraction of the genetic variability in a quantitative trait. Unlike simple interval mapping, composite interval mapping usually controls for variation produced at one or more background marker loci. These background markers are generally chosen because they are already known to be close to the location of a significant QTL. By factoring out a portion of the genetic variance produced by a major QTL, one can occasionally detect secondary QTLs. WebQTL allows users to control for a single background marker. To select this marker, first run the **Marker Regression** analysis (and if necessary, check the box labeled display all LRS, select the appropriate locus, and the click on either **Composite Interval Mapping** or **Composite Regression**. A more powerful and effective alternative to composite interval mapping is pair-scan analysis. This latter method takes into accounts (models) both the independent effects of two loci and possible two-locus epistatic interactions. [Williams RW, Dec 20, 2004] <div id="Correlations"></div> #### Correlations: Pearson and Spearman: -GeneNetwork provides tools to compute both Pearson product-moment correlations (the standard type of correlation), Spearman rank order correlations. [Wikipedia](http://en.wikipedia.org/wiki/Pearson_product-moment_correlation_coefficient) and introductory statistics text will have a discussion of these major types of correlation. The quick advice is to use the more robust Spearman rank order correlation if the number of pairs of observations in a data set is less than about 30 and to use the more powerful but much more sensitive Pearson product-moment correlation when the number of observations is greater than 30 AND after you have dealt with any outliers. GeneNetwork automatically flags outliers for you in the Trait Data and Analysis form. GeneNetwork also allows you to modify values by either deleting or winsorising them. That means that you can use Pearson correlations even with smaller sample sizes after making sure that data are well distributed. Be sure to view the scatterplots associated with correlation values (just click on the value to generate a plot). Look for bivariate outliers. +GeneNetwork provides tools to compute both Pearson product-moment correlations (the standard type of correlation), Spearman rank order correlations. [Wikipedia](http://en.wikipedia.org/wiki/Pearson_product-moment_correlation_coefficient) and introductory statistics text will have a discussion of these major types of correlation. The quick advice is to use the more robust Spearman rank order correlation if the number of pairs of observations in a data set is less than about 30 and to use the more powerful but much more sensitive Pearson product-moment correlation when the number of observations is greater than 30 AND after you have dealt with any outliers. GeneNetwork automatically flags outliers for you in the Trait Data and Analysis form. GeneNetwork also allows you to modify values by either deleting or winsorising them. That means that you can use Pearson correlations even with smaller sample sizes after making sure that data are well distributed. Be sure to view the scatterplots associated with correlation values (just click on the value to generate a plot). Look for bivariate outliers. #### Cross: -The term Cross refers to a group of offspring made by mating (crossing) one strain with another strain. There are several types of crosses including intercrosses, backcrosses, advanced intercrosses, and recombinant inbred intercrosses. Genetic crosses are almost always started by mating two different but fully inbred strains to each other. For example, a B6D2F2 cross is made by breeding C57BL/6J females (B6 or B for short) with DBA/2J males (D2 or D) and then intercrossing their F1 progeny to make the second filial generation (F2). By convention the female is always listed first in cross nomenclature; B6D2F2 and D2B6F2 are therefore so-called reciprocal F2 intercrosses (B6D2F1 females to B6D2F1 males or D2B6F1 females to D2B6F1 males). A cross may also consist of a set of recombinant inbred (RI) strains such as the BXD strains, that are actually inbred progeny of a set of B6D2F2s. Crosses can be thought of as a method to randomize the assignment of blocks of chromosomes and genetic variants to different individuals or strains. This random assignment is a key feature in testing for causal relations. The strength with which one can assert that a causal relation exists between a chromosomal location and a phenotypic variant is measured by the LOD score or the LRS score (they are directly convertable, where LOD = LRS/4.61) [Williams RW, Dec 26, 2004; Dec 4, 2005]. - +The term Cross refers to a group of offspring made by mating (crossing) one strain with another strain. There are several types of crosses including intercrosses, backcrosses, advanced intercrosses, and recombinant inbred intercrosses. Genetic crosses are almost always started by mating two different but fully inbred strains to each other. For example, a B6D2F2 cross is made by breeding C57BL/6J females (B6 or B for short) with DBA/2J males (D2 or D) and then intercrossing their F1 progeny to make the second filial generation (F2). By convention the female is always listed first in cross nomenclature; B6D2F2 and D2B6F2 are therefore so-called reciprocal F2 intercrosses (B6D2F1 females to B6D2F1 males or D2B6F1 females to D2B6F1 males). A cross may also consist of a set of recombinant inbred (RI) strains such as the BXD strains, that are actually inbred progeny of a set of B6D2F2s. Crosses can be thought of as a method to randomize the assignment of blocks of chromosomes and genetic variants to different individuals or strains. This random assignment is a key feature in testing for causal relations. The strength with which one can assert that a causal relation exists between a chromosomal location and a phenotypic variant is measured by the LOD score or the LRS score (they are directly convertable, where LOD = LRS/4.61) [Williams RW, Dec 26, 2004; Dec 4, 2005]. + [Go back to index](#index) <div id="d"></div> @@ -89,15 +89,15 @@ The term dominance indicates that the phenotype of intercross progeny closely re The dominance effects are computed at each location on the maps generated by the WebQTL module for F2 populations (e.g., B6D2F2 and B6BTBRF2). Orange and purple line colors are used to distinguish the polarity of the dominance effects. Purple is the positive dominance effect that matches the polarity of the green additive effect, whereas orange is the negative dominance effect that matches the polarity of the red additive effect. -Note that dominance deviations cannot be computed from a set of recombinant inbred strains because there are only two classes of genotypes at any marker (aa and AA, more usuually written AA and BB). However, when data for F1 hybrids are available one can estimate the dominance of the trait. This global phenotypic dominance has almost nothing to do with the dominance deviation at a single marker in the genome. In other words, the dominance deviation detected at a single marker may be reversed or neutralized by the action of many other polymorphic genes. [Williams RW, Dec 21, 2004; Sept 3, 2005] - +Note that dominance deviations cannot be computed from a set of recombinant inbred strains because there are only two classes of genotypes at any marker (aa and AA, more usuually written AA and BB). However, when data for F1 hybrids are available one can estimate the dominance of the trait. This global phenotypic dominance has almost nothing to do with the dominance deviation at a single marker in the genome. In other words, the dominance deviation detected at a single marker may be reversed or neutralized by the action of many other polymorphic genes. [Williams RW, Dec 21, 2004; Sept 3, 2005] + [Go back to index](#index) <div id="e"></div> #### Epistasis: -Epistasis means that combined effects of two or more different loci or polymorphic genes are not what one would expect given the addition of their individual effects. There is, in other words, evidence for non-linear interactions among two or more loci. This is similar to the dominance effects mentioned above, but now generalized to two or more distinct loci, rather than to two or more alleles at a single locus. For example, if QTL 1 has an A allele that has an additive effects of +5 and QTL 2 has an A alleles that has an additive effect of +2, then the two locus genotype combination A/A would be expected to boost the mean by +7 units. But if the value of these A/A individuals was actually -7 we would be quite surprised and would refer to this as an epistatic interaction between QTL 1 and QTL 2. WebQTL will search for all possible epistatic interactions between pairs of loci in the genome. This function is called a **Pair Scan** becasue the software analyzes the LRS score for all possible pairs of loci. Instead of viewing an LRS plot along a single dimension, we now view a two-dimensional plot that shows a field of LRS scores computed for pairs of loci. Pair scan plots are extremely sensitive to outlier data. Be sure to review the primary data carefully using **Basic Statistics**. Also note that this more sophisiticated method also demands a significantly larger sample size. While 25 to 50 cases may be adequate for a conventional LRS plot (sometimes called a "main scan"), a **Pair-Scan** is hard to apply safely with fewer than 60 cases. [Williams RW, Dec 21, 2004; Dec 5, 2005] +Epistasis means that combined effects of two or more different loci or polymorphic genes are not what one would expect given the addition of their individual effects. There is, in other words, evidence for non-linear interactions among two or more loci. This is similar to the dominance effects mentioned above, but now generalized to two or more distinct loci, rather than to two or more alleles at a single locus. For example, if QTL 1 has an A allele that has an additive effects of +5 and QTL 2 has an A alleles that has an additive effect of +2, then the two locus genotype combination A/A would be expected to boost the mean by +7 units. But if the value of these A/A individuals was actually -7 we would be quite surprised and would refer to this as an epistatic interaction between QTL 1 and QTL 2. WebQTL will search for all possible epistatic interactions between pairs of loci in the genome. This function is called a **Pair Scan** becasue the software analyzes the LRS score for all possible pairs of loci. Instead of viewing an LRS plot along a single dimension, we now view a two-dimensional plot that shows a field of LRS scores computed for pairs of loci. Pair scan plots are extremely sensitive to outlier data. Be sure to review the primary data carefully using **Basic Statistics**. Also note that this more sophisiticated method also demands a significantly larger sample size. While 25 to 50 cases may be adequate for a conventional LRS plot (sometimes called a "main scan"), a **Pair-Scan** is hard to apply safely with fewer than 60 cases. [Williams RW, Dec 21, 2004; Dec 5, 2005] #### Effect Size of a QTL: @@ -108,20 +108,20 @@ Please note that the functional importance of a locus, QTL, or GWAS hit can not Estimates of effect size for families of inbred lines, such as the BXD, HXB, CC, and hybrid diversity panels (e.g. the hybrid mouse diversity panel and the hybrid rat diversity panel) are typically (and correctly) much higher than those measured in otherwise similar analysis of intercrosses, heterogeneous stock (HS), or diversity outbred stock. Two factors contribute to the much higher level of explained variance of QTLs when using inbred strain panels. -1. **Replication Rate:** The variance that can be explained by a locus is increased by sampling multiple cases that have identical genomes and by using the strain mean for genetic analysis. Increasing replication rates from 1 to 6 can easily double the apparent heritability of a trait and therefore the effect size of a locus. The reason is simple???resampling decrease the standard error of mean, boosting the effective heritability (see Glossary entry on *Heritability* and focus on figure 1 from the Belknap [1998](http://gn1.genenetwork.org/images/upload/Belknap_Heritability_1998.pdf) paper reproduced below).<br/>Compare the genetically explained variance (labeled h2RI in this figure) of a single case (no replication) on the x-axis with the function at a replication rate of 4 on the y-axis. If the explained variance is 0.1 (10% of all variance explained) then the value is boosted to 0.3 (30% of strain mean variance explained) with n = 4. +1. **Replication Rate:** The variance that can be explained by a locus is increased by sampling multiple cases that have identical genomes and by using the strain mean for genetic analysis. Increasing replication rates from 1 to 6 can easily double the apparent heritability of a trait and therefore the effect size of a locus. The reason is simple--resampling decrease the standard error of mean, boosting the effective heritability (see Glossary entry on *Heritability* and focus on figure 1 from the Belknap [1998](http://gn1.genenetwork.org/images/upload/Belknap_Heritability_1998.pdf) paper reproduced below).<br/>Compare the genetically explained variance (labeled h2RI in this figure) of a single case (no replication) on the x-axis with the function at a replication rate of 4 on the y-axis. If the explained variance is 0.1 (10% of all variance explained) then the value is boosted to 0.3 (30% of strain mean variance explained) with n = 4. 2. **Homozygosity:** The second factor has to do with the inherent genetic variance of populations. Recombinant inbred lines are homozygous at nearly all loci. This doubles the genetic variance in a family of recombinant inbred lines compared to a matched number of F2s. This also quadruples the variance compared to a matched number of backcross cases. As a result 40 BXDs sampled just one per genometype will average 2X the genetic variance and 2X the heritability of 40 BDF2 cases. Note that panels made up of isogenic F1 hybrids (so-called diallel crosses, DX) made by crossing recombinant inbred strains (BXD, CC, or HXB) are no longer homozygous at all loci, and while they do expose important new sources of variance associated with dominance, they do not benefit from the 2X gain in genetic variance relative to an F2 intercross. <img width="600px" src="/static/images/Belknap_Fig1_1998.png" alt="Homozygosity" /> -For the reasons listed above a QTL effect size of 0.4 detected a panel of BXD lines replicated four times each (160 cases total), corresponds approximately to an effect size of 0.18 in BXDs without replication (40 cases total), and to an effect size of 0.09 in an F2 of 40 cases total. [Williams RW, Dec 23, 2004; updated by RWW July 13, 2019] +For the reasons listed above a QTL effect size of 0.4 detected a panel of BXD lines replicated four times each (160 cases total), corresponds approximately to an effect size of 0.18 in BXDs without replication (40 cases total), and to an effect size of 0.09 in an F2 of 40 cases total. [Williams RW, Dec 23, 2004; updated by RWW July 13, 2019] #### eQTL, cis eQTL, trans eQTL An expression QTL or eQTL. Differences in the expression of mRNA or proteins are often treated as standard phenotypes, much like body height or lung capacity. The variation in these microscopic traits (microtraits) can be mapped using conventional QTL methods. [Damerval](http://www.genetics.org/cgi/reprint/137/1/289) and colleagues were the first authors to use this kind of nomenclature and in their classic study of 1994 introduced the term PQLs for protein quantitative trait loci. Schadt and colleagues added the acronym eQTL in their early mRNA study of corn, mouse, and humans. We now are "blessed" with all kinds of prefixes to QTLs that highlight the type of trait that has been measured (m for metabolic, b for behavioral, p for physiological or protein). -eQTLs of mRNAs and proteins have the unique property of (usually) having a single parent gene and genetic location. An eQTL that maps to the location of the parent gene that produces the mRNA or protein is referred to as a **cis eQTL** or local eQTL. In contrast, an eQTL that maps far away from its parent gene is referred to as a **trans eQTL**. You can use special search commands in GeneNetwork to find cis and trans eQTLs. [Williams RW, Nov 23, 2009, Dec 2009] - +eQTLs of mRNAs and proteins have the unique property of (usually) having a single parent gene and genetic location. An eQTL that maps to the location of the parent gene that produces the mRNA or protein is referred to as a **cis eQTL** or local eQTL. In contrast, an eQTL that maps far away from its parent gene is referred to as a **trans eQTL**. You can use special search commands in GeneNetwork to find cis and trans eQTLs. [Williams RW, Nov 23, 2009, Dec 2009] + [Go back to index](#index) <div id="f"></div> @@ -130,12 +130,12 @@ eQTLs of mRNAs and proteins have the unique property of (usually) having a singl #### Frequency of Peak LRS: -The height of the yellow bars in some of the Map View windows provides a measure of the confidence with which a trait maps to a particular chromosomal region. WebQTL runs 2000 bootstrap samples of the original data. (A bootstrap sample is a "sample with replacement" of the same size as the original data set in which some samples will by chance be represented one of more times and others will not be represented at all.) For each of these 2000 bootstraps, WebQTL remaps each and keeps track of the location of the single locus with the highest LRS score. These accumulated locations are used to produce the yellow histogram of "best locations." A frequency of 10% means that 200 of 2000 bootstraps had a peak score at this location. It the mapping data are robust (for example, insensitive to the exclusion of an particular case), then the bootstrap bars should be confined to a short chromosomal interval. Bootstrap results will vary slightly between runs due to the random generation of the bootstrap samples. [Williams RW, Oct 15, 2004] +The height of the yellow bars in some of the Map View windows provides a measure of the confidence with which a trait maps to a particular chromosomal region. WebQTL runs 2000 bootstrap samples of the original data. (A bootstrap sample is a "sample with replacement" of the same size as the original data set in which some samples will by chance be represented one of more times and others will not be represented at all.) For each of these 2000 bootstraps, WebQTL remaps each and keeps track of the location of the single locus with the highest LRS score. These accumulated locations are used to produce the yellow histogram of "best locations." A frequency of 10% means that 200 of 2000 bootstraps had a peak score at this location. It the mapping data are robust (for example, insensitive to the exclusion of an particular case), then the bootstrap bars should be confined to a short chromosomal interval. Bootstrap results will vary slightly between runs due to the random generation of the bootstrap samples. [Williams RW, Oct 15, 2004] #### False Discovery Rate (FDR): -A [false discovery](http://en.wikipedia.org/wiki/False_discovery_rate) is an apparently significant finding--usually determined using a particular P value alpha criterion--that given is known to be insignificant or false given other information. When performing a single statistical test we often accept a false discovery rate of 1 in 20 (p = .05). False discovery rates can climb to high levels in large genomic and genetic studies in which hundreds to millions of tests are run and summarized using standard "single test" p values. There are various statistical methods to estimate and control false discovery rate and to compute genome-wide p values that correct for large numbers of implicit or explicit statistical test. The Permutation test in GeneNetwork is one method that is used to prevent and excessive number of false QTL discoveries. Methods used to correct the FDR are approximations and may depend on a set of assumptions about data and sample structure. [Williams RW, April 5, 2008] - +A [false discovery](http://en.wikipedia.org/wiki/False_discovery_rate) is an apparently significant finding--usually determined using a particular P value alpha criterion--that given is known to be insignificant or false given other information. When performing a single statistical test we often accept a false discovery rate of 1 in 20 (p = .05). False discovery rates can climb to high levels in large genomic and genetic studies in which hundreds to millions of tests are run and summarized using standard "single test" p values. There are various statistical methods to estimate and control false discovery rate and to compute genome-wide p values that correct for large numbers of implicit or explicit statistical test. The Permutation test in GeneNetwork is one method that is used to prevent and excessive number of false QTL discoveries. Methods used to correct the FDR are approximations and may depend on a set of assumptions about data and sample structure. [Williams RW, April 5, 2008] + [Go back to index](#index) <div id="g"></div> @@ -146,7 +146,7 @@ A [false discovery](http://en.wikipedia.org/wiki/False_discovery_rate) is an app GeneNetwork provides summary information on most of the genes and their transcripts. Genes and their alternative splice variants are often are poorly annotated and may not have proper names or symbols. However, almost all entries have a valid GenBank accession identifier. This is a unique code associated with a single sequence deposited in GenBank (Entrez Nucleotide). A single gene may have hundreds of GenBank entries. GenBank entries that share a genomic location and possibly a single gene are generally combined into a single UniGene entry. For mouse, these always begin with "Mm" (Mus musculus) and are followed by a period and then a number. More than half of all mouse UniGene identifiers are associated with a reputable gene, and these genes will have gene identifiers (GeneID). GeneIDs are identical to LocusLink identifiers (LocusID). Even a 10 megabase locus such as human Myopia 4 (MYP4) that is not yet associated with a specific gene is assigned a GeneID--a minor misnomer and one reason to prefer the term LocusID. -See the related [FAQ](http://gn1.genenetwork.org/faq.html#Q-6) on "How many genes and transcripts are in your databases and what fraction of the genome is being surveyed?" [Williams RW, Dec 23, 2004, updated Jan 2, 2005] +See the related [FAQ](http://gn1.genenetwork.org/faq.html#Q-6) on "How many genes and transcripts are in your databases and what fraction of the genome is being surveyed?" [Williams RW, Dec 23, 2004, updated Jan 2, 2005] #### Genetic Reference Population (GRP): @@ -162,19 +162,19 @@ There are several subtypes of GRPs. In addition to recombinant inbred strains th - Recombinant congenic ([RCC](http://research.jax.org/grs/type/recombcong.htmll)) strains such as the [AcB](http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=11374899&query_hl=4) set Consomic or chromosome substitution strains ([CSS](http://research.jax.org/grs/type/consomic.html)) of mice (Matin et al., [1999](http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=10508525&query_hl=11)) and rats (Roman et al., [2002](http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=12858554&query_hl=7)) - + - Recombinant intercross ([RIX](http://www.ncbi.nlm.nih.gov/pubmed/?term=15879512)) F1 sets made by mating different RI strains to each other to generate large set of R! first generation (F1) progeny (RIX). This is a standard ([diallel cross](http://en.wikipedia.org/wiki/Diallel_cross)) of RI inbred strains. Genetic analysis of a set of RIX progeny has some advantages over a corresponding analysis of RI strains. The first of these is that while each set of F1 progeny is fully isogenic (AXB1 x AXB2 gives a set of isogenic F1s), these F1s are not inbred but are heterozygous at many loci across the genome. RIX therefore retain the advance of being genetically defined and replicable, but without the disadvantage of being fully inbred. RIX have a genetic architecture more like natural populations. The second correlated advantage is that it is possible to study patterns of dominance of allelic variants using an RIX cross. Almost all loci or genes that differs between the original stock strains (A and B) will be heterozygous among a sufficiently larges set of RIX. A set of RIX progeny can therefore be mapped using the same methods used to map an F2 intercross. Mapping of QTLs may have somewhat more power and precision than when RI strains are used alone. A third advantage is that RIX sets make it possible to expand often limited RI resources to very large sizes to confirm and extend models of genetic or GXE effects. For example a set of 30 AXB strains can be used to generate a full matrix of 30 x 29 unique RIX progeny. The main current disadvantage of RIX panels is the comparative lack of extant phenotype data. - -- Recombinant F1 line sets can also be made by backcrossing an entire RI sets to a single inbred line that carries an interesting mutation or transgene (RI backcross or RIB). GeneNetwork includes one RI backcross sets generated by Kent Hunter. In this RIB each of 18 AKXD RI strains were crossed to an FVB/N line that carries a tumor susceptibility allele (polyoma middle T). - + +- Recombinant F1 line sets can also be made by backcrossing an entire RI sets to a single inbred line that carries an interesting mutation or transgene (RI backcross or RIB). GeneNetwork includes one RI backcross sets generated by Kent Hunter. In this RIB each of 18 AKXD RI strains were crossed to an FVB/N line that carries a tumor susceptibility allele (polyoma middle T). + All of these sets of lines are GRPs since each line is genetically defined and because the set as a whole can in principle be easily regenerated and phenotyped. Finally, each of these resources can be used to track down genetic loci that are causes of variation in phenotype using variants of standard linkage analysis. A Diversity Panel such as that used by the Mouse Phenome Project is not a standard GRPs, although its also shares the ability to accumulate and study networks of phenotypes. The main difference is that a Diversity Panel cannot be used for conventional linkage analysis. A sufficiently large Diversity Panel could in principle be used for the equivalent of an assocation study. However, these are definitely NOT in silico studies, because hundreds of individuals need to be phenotyped for every trait. Surveys of many diverse isogenic lines (inbred or F1 hybrids) is statistically the equivalent of a human association study (the main difference is the ability to replicate measurements and study sets of traits) and therefore, like human association studies, does require very high sample size to map polygenic traits. Like human association studies there is also a high risk of false positive results due to population stratification and non-syntenic marker association. -A good use of a Diversity Panel is as a fine-mapping resource with which to dissect chromosomal intervals already mapped using a conventional cross or GRP. GeneNetwork now includes Mouse Diversity Panel (MDP) data for several data sets. We now typically include all 16 sequenced strains of mice, and add PWK/PhJ, NZO/HiLtJ (two of the eight members of the Collaborative Cross), and several F1 hybrids. The MDP data is often appended at the bottom of the GRP data set with which is was acquired (e.g., BXD hippocampal and BXD eye data sets). [Williams RW, June 19, 2005; Dec 4, 2005] +A good use of a Diversity Panel is as a fine-mapping resource with which to dissect chromosomal intervals already mapped using a conventional cross or GRP. GeneNetwork now includes Mouse Diversity Panel (MDP) data for several data sets. We now typically include all 16 sequenced strains of mice, and add PWK/PhJ, NZO/HiLtJ (two of the eight members of the Collaborative Cross), and several F1 hybrids. The MDP data is often appended at the bottom of the GRP data set with which is was acquired (e.g., BXD hippocampal and BXD eye data sets). [Williams RW, June 19, 2005; Dec 4, 2005] #### Genotype - + The state of a gene or DNA sequence, usually used to describe a contrast between two or more states, such as that between the normal state (wildtype) and a mutant state (mutation) or between the alleles inherited from two parents. All species that are included in GeneNetwork are diploid (derived from two parents) and have two copies of most genes (genes located on the X and Y chromosomes are exceptions). As a result the genotype of a particular diploid individual is actually a pair of genotypes, one from each parents. For example, the offspring of a mating between strain A and strain B will have one copy of the A genotype and one copy of the B genotype and therefore have an A/B genotype. In contrast, offspring of a mating between a female strain A and a male strain A will inherit only A genotypes and have an A/A genotype. Genotypes can be measured or inferred in many different ways, even by visual inspection of animals (e.g. as Gregor Mendel did long before DNA was discovered). But now the typical method is to directly test DNA that has a well define chromosomal location that has been obtained from one or usually many cases using molecular tests that often rely on polymerase chain reaction steps and sequence analysis. Each case is genotyped at many chromosomal locations (loci, markers, or genes). The entire collection of genotypes (as many a 1 million for a single case) is also sometimes referred to as the cases genotype, but the word "genometype" might be more appropriate to highlight the fact that we are now dealing with a set of genotypes spanning the entire genome (all chromosomes) of the case. @@ -182,7 +182,7 @@ Genotypes can be measured or inferred in many different ways, even by visual ins For gene mapping purposes, genotypes are often translated from letter codes (A/A, A/B, and B/B) to simple numerical codes that are more suitable for computation. A/A might be represented by the value -1, A/B by the value 0, and B/B by the value +1. This recoding makes it easy to determine if there is a statistically significant correlation between genotypes across of a set of cases (for example, an F2 population or a Genetic Reference Panel) and a variable phenotype measured in the same population. A sufficiently high correlation between genotypes and phenotypes is referred to as a quantitative trait locus (QTL). If the correlation is almost perfect (r > 0.9) then correlation is usually referred to as a Mendelian locus. Despite the fact that we use the term "correlation" in the preceding sentences, the genotype is actually the cause of the phenotype. More precisely, variation in the genotypes of individuals in the sample population cause the variation in the phenotype. The statistical confidence of this assertion of causality is often estimated using LOD and LRS scores and permutation methods. If the LOD score is above 10, then we can be extremely confident that we have located a genetic cause of variation in the phenotype. While the location is defined usually with a precision ranging from 10 million to 100 thousand basepairs (the locus), the individual sequence variant that is responsible may be quite difficult to extract. Think of this in terms of police work: we may know the neighborhood where the suspect lives, we may have clues as to identity and habits, but we still may have a large list of suspects. Text here [Williams RW, July 15, 2010] - + [Go back to index](#index) <div id="h"></div> @@ -193,7 +193,7 @@ Text here [Williams RW, July 15, 2010] Heritability is a rough measure of the ability to use genetic information to predict the level of variation in phenotypes among progeny. Values range from 0 to 1 (or 0 to 100%). A value of 1 or 100% means that a trait is entirely predictable based on paternal and maternal genetic information (for example, a strong Mendelian trait), whereas a value of 0 means that a trait is not at all predictable from information on gene variants. Estimates of heritability are highly dependent on the environment, stage, and age. -Important traits that affect fitness often have low heritabilities because stabilizing selection reduces the frequency of DNA variants that produce suboptimal phenotypes. Conversely, less critical traits for which substantial phenotypic variation is well tolerated, may have high heritability. The environment of laboratory rodents is unnatural, and this allows the accumulation of somewhat deleterious mutations (for example, mutations that lead to albinism). This leads to an upward trend in heritability of unselected traits in laboratory populations--a desirable feature from the point of view of the biomedical analysis of the genetic basis of trait variance. Heritability is a useful parameter to measure at an early stage of a genetic analysis, because it provides a rough gauge of the likelihood of successfully understanding the allelic sources of variation. Highly heritable traits are more amenable to mapping studies. There are numerous ways to estimate heritability, a few of which are described below. [Williams RW, Dec 23, 2004] +Important traits that affect fitness often have low heritabilities because stabilizing selection reduces the frequency of DNA variants that produce suboptimal phenotypes. Conversely, less critical traits for which substantial phenotypic variation is well tolerated, may have high heritability. The environment of laboratory rodents is unnatural, and this allows the accumulation of somewhat deleterious mutations (for example, mutations that lead to albinism). This leads to an upward trend in heritability of unselected traits in laboratory populations--a desirable feature from the point of view of the biomedical analysis of the genetic basis of trait variance. Heritability is a useful parameter to measure at an early stage of a genetic analysis, because it provides a rough gauge of the likelihood of successfully understanding the allelic sources of variation. Highly heritable traits are more amenable to mapping studies. There are numerous ways to estimate heritability, a few of which are described below. [Williams RW, Dec 23, 2004] #### h<sup>2</sup> Estimated by Intraclass Correlation: @@ -201,7 +201,7 @@ Heritability can be estimated using the intraclass correlation coefficient. This <math>r = (Between-strain MS - Within-strain MS)/(Between-strain MS + (n-1)x Within-strain MS)</math> -where n is the average number of cases per strain. The intraclass correlation approaches 1 when there is minimal variation within strains, and strain means differ greatly. In contrast, if difference between strains are less than what would be predicted from the differences within strain, then the intraclass correlation will produce negative estimates of heritability. Negative heritability is usually a clue that the design of the experiment has injected excessive within-strain variance. It is easy for this to happen inadvertently by failing to correct for a batch effect. For example, if one collects the first batch of data for strains 1 through 20 during a full moon, and a second batch of data for these same strains during a rare blue moon, then the apparent variation within strain may greatly exceed the among strain variance. A technical batch effect has been confounded with the within-strain variation and has swamped any among-strain variance. What to do? Fix the batch effect, sex effect, age effect, etc., first! [Williams RW, Chesler EJ, Dec 23, 2004] +where n is the average number of cases per strain. The intraclass correlation approaches 1 when there is minimal variation within strains, and strain means differ greatly. In contrast, if difference between strains are less than what would be predicted from the differences within strain, then the intraclass correlation will produce negative estimates of heritability. Negative heritability is usually a clue that the design of the experiment has injected excessive within-strain variance. It is easy for this to happen inadvertently by failing to correct for a batch effect. For example, if one collects the first batch of data for strains 1 through 20 during a full moon, and a second batch of data for these same strains during a rare blue moon, then the apparent variation within strain may greatly exceed the among strain variance. A technical batch effect has been confounded with the within-strain variation and has swamped any among-strain variance. What to do? Fix the batch effect, sex effect, age effect, etc., first! [Williams RW, Chesler EJ, Dec 23, 2004] #### h<sup>2</sup> Estimated using Hegmann and Possidente's Method (Adjusted Heritability in the Basic Statisics): @@ -215,12 +215,12 @@ However, this estimate of h2 cannot be compared directly to those calculated usi <math>h<sup>2</sup> = 0.5Va / (0.5Va+Ve)</math> -The factor 0.5 is applied to Va to adjust for the overestimation of additive genetic variance among inbred strains. This estimate of heritability also does not make allowances for the within-strain error term. The 0.5 adjustment factor is not recommended any more because h2 is severely **underestimated**. This adjustment is really only needed if the goal is to compare h2 between intercrosses and those generated using panels of inbred strains. +The factor 0.5 is applied to Va to adjust for the overestimation of additive genetic variance among inbred strains. This estimate of heritability also does not make allowances for the within-strain error term. The 0.5 adjustment factor is not recommended any more because h2 is severely **underestimated**. This adjustment is really only needed if the goal is to compare h2 between intercrosses and those generated using panels of inbred strains. -#### h<sup>2</sup>RIx?? +#### h<sup>2</sup>RIx -Finally, heritability calculations using strain means, such as those listed above, do not provide estimates of the effective heritability achieved by resampling a given line, strain, or genometype many times. Belknap ([1998](http://gn1.genenetwork.org/images/upload/Belknap_Heritability_1998.pdf)) provides corrected estimates of the effective heritability. Figure 1 from his paper (reproduced below) illustrates how resampling helps a great deal. Simply resampling each strain 8 times can boost the effective heritability from 0.2 to 0.8. The graph also illustrates why it often does not make sense to resample much beyond 4 to 8, depending on heritability. Belknap used the term h2RIx?? in this figure and paper, since he was focused on data generated using recombinant inbred (RI) strains, but the logic applies equally well to any panel of genomes for which replication of individual genometypes is practical. This h2RIx?? can be calculated simply by: -<math>h<sup>2</sup><sub>RIx??</sub> = V<sub>a</sub> / (V<sub>a</sub>+(V<sub>e</sub>/n))</math> where V<sub>a</sub> is the genetic variability (variability between strains), V<sub>e</sub> is the environmental variability (variability within strains), and n is the number of within strain replicates. Of course, with many studies the number of within strain replicates will vary between strains, and this needs to be dealt with. A reasonable approach is to use the harmonic mean of n across all strains. +Finally, heritability calculations using strain means, such as those listed above, do not provide estimates of the effective heritability achieved by resampling a given line, strain, or genometype many times. Belknap ([1998](http://gn1.genenetwork.org/images/upload/Belknap_Heritability_1998.pdf)) provides corrected estimates of the effective heritability. Figure 1 from his paper (reproduced below) illustrates how resampling helps a great deal. Simply resampling each strain 8 times can boost the effective heritability from 0.2 to 0.8. The graph also illustrates why it often does not make sense to resample much beyond 4 to 8, depending on heritability. Belknap used the term h2RIx in this figure and paper, since he was focused on data generated using recombinant inbred (RI) strains, but the logic applies equally well to any panel of genomes for which replication of individual genometypes is practical. This h2RIx can be calculated simply by: +<math>h<sup>2</sup><sub>RIx</sub> = V<sub>a</sub> / (V<sub>a</sub>+(V<sub>e</sub>/n))</math> where V<sub>a</sub> is the genetic variability (variability between strains), V<sub>e</sub> is the environmental variability (variability within strains), and n is the number of within strain replicates. Of course, with many studies the number of within strain replicates will vary between strains, and this needs to be dealt with. A reasonable approach is to use the harmonic mean of n across all strains. <img width="600px" src="/static/images/Belknap_Fig1_1998.png" alt="Homozygosity" /> @@ -228,7 +228,7 @@ An analysis of statistical power is useful to estimate numbers of replicates and **[Power Calculator (D. Ashbrook)](https://dashbrook1.shinyapps.io/bxd_power_calculator_app/)** -We can see that in all situations power is increased more by increasing the number of lines than by increasing the number of biological replicates. Dependent upon the heritability of the trait, there is little gain in power when going above 4-6 biological replicates. [DGA, Feb 1, 2019] [Chesler EJ, Dec 20, 2004; RWW updated March 7, 2018; Ashbrook DG, updated Feb 1, 2019] +We can see that in all situations power is increased more by increasing the number of lines than by increasing the number of biological replicates. Dependent upon the heritability of the trait, there is little gain in power when going above 4-6 biological replicates. [DGA, Feb 1, 2019] [Chesler EJ, Dec 20, 2004; RWW updated March 7, 2018; Ashbrook DG, updated Feb 1, 2019] #### Hitchhiking Effect: @@ -236,7 +236,7 @@ Conventional knockout lines (KOs) of mice are often mixtures of the genomes of t **Genetics of KO Lines**. The embryonic stem cells used to make KOs are usually derived from a 129 strain of mouse (e.g., 129/OlaHsd). Mutated stem cells are then added to a C57BL/6J blastocyst to generate B6x129 chimeric mice. Germline transmission of the KO allele is tested and carriers are then used to establish heterozygous +/- B6.129 KO stock. This stock is often crossed back to wildtype C57BL/6J strains for several generations. At each generation the transmission of the KO is checked by genotyping the gene or closely flanking markers in each litter of mice. Carriers are again selected for breeding. The end result of this process is a KO congenic line in which the genetic background is primarily C57BL/6J except for the region around the KO gene. -It is often thought that 10 generations of backcrossing will result in a pure genetic background (99.8% C57BL/6J). Unfortunately, this is not true for the region around the KO, and even after many generations of backcrossing of KO stock to C57BL/6J, a large region around the KO is still derived from the 129 substrain (see the residual white "line" at N10 in the figure below. +It is often thought that 10 generations of backcrossing will result in a pure genetic background (99.8% C57BL/6J). Unfortunately, this is not true for the region around the KO, and even after many generations of backcrossing of KO stock to C57BL/6J, a large region around the KO is still derived from the 129 substrain (see the residual white "line" at N10 in the figure below. <img width="300px" src="/static/images/Congenic.png" alt="Congenic" /> @@ -251,8 +251,8 @@ What is the solution? 2. Use a KO in which the KO has been backcrossed to a 129 strain--ideally the same strain from which ES cells were obtained. This eliminates the hitchhiker effect entirely and the KO, HET, and WT littermates really can be compared. 3. Use a conditional KO. - -4. Compare the phenotype of the two parental strains--129 and C57BL/6J and see if they differ in ways that might be confounded with the effects of the KO. + +4. Compare the phenotype of the two parental strains--129 and C57BL/6J and see if they differ in ways that might be confounded with the effects of the KO. <img width="600px" src="/static/images/SilverFig3_6.png" alt="Homozygosity" /> @@ -270,7 +270,7 @@ The interquartile range is the difference between the 75% and 25% percentiles of #### Interval Mapping: -Interval mapping is a process in which the statistical significance of a hypothetical QTL is evaluated at regular points across a chromosome, even in the absence of explicit genotype data at those points. In the case of WebQTL, significance is calculated using an efficient and very rapid regression method, the Haley-Knott regression equations ([Haley CS, Knott SA. 1992. A simple regression method for mapping quantitative trait loci in line crosses using flanking markers; Heredity 69:315???324](http://www.ncbi.nlm.nih.gov/pubmed/16718932)), in which trait values are compared to the known genotype at a marker or to the probability of a specific genotype at a test location between two flanking markers. (The three genotypes are coded as -1, 0, and +1 at known markers, but often have fractional values in the intervals between markers.) The inferred probability of the genotypes in regions that have not been genotyped can be estimated from genotypes of the closest flanking markers. GeneNetwork/WebQTL compute linkage at intervals of 1 cM or less. As a consequence of this approach to computing linkage statistics, interval maps often have a characteristic shape in which the markers appear as sharply defined inflection points, and the intervals between nodes are smooth curves. [Chesler EJ, Dec 20, 2004; RWW April 2005; RWW Man 2014] +Interval mapping is a process in which the statistical significance of a hypothetical QTL is evaluated at regular points across a chromosome, even in the absence of explicit genotype data at those points. In the case of WebQTL, significance is calculated using an efficient and very rapid regression method, the Haley-Knott regression equations ([Haley CS, Knott SA. 1992. A simple regression method for mapping quantitative trait loci in line crosses using flanking markers; Heredity 69:315-324](http://www.ncbi.nlm.nih.gov/pubmed/16718932)), in which trait values are compared to the known genotype at a marker or to the probability of a specific genotype at a test location between two flanking markers. (The three genotypes are coded as -1, 0, and +1 at known markers, but often have fractional values in the intervals between markers.) The inferred probability of the genotypes in regions that have not been genotyped can be estimated from genotypes of the closest flanking markers. GeneNetwork/WebQTL compute linkage at intervals of 1 cM or less. As a consequence of this approach to computing linkage statistics, interval maps often have a characteristic shape in which the markers appear as sharply defined inflection points, and the intervals between nodes are smooth curves. [Chesler EJ, Dec 20, 2004; RWW April 2005; RWW Man 2014] #### Interval Mapping Options: @@ -286,8 +286,8 @@ Interval mapping is a process in which the statistical significance of a hypothe - _Display from X Mb to Y Mb_: Enter values in megabases to regenerate a smaller or large map view. -- _Graph width (in pixels)_: Adjust this value to obtain larger or smaller map views (x axis only). - +- _Graph width (in pixels)_: Adjust this value to obtain larger or smaller map views (x axis only). + [Go back to index](#index) <div id="j"></div> @@ -310,9 +310,9 @@ Interval mapping is a process in which the statistical significance of a hypothe #### Literature Correlation: -The literature correlation is a unique feature in GeneNetwork that quantifies the similarity of words used to describe genes and their functions. Sets of words associated with genes were extracted from MEDLINE/PubMed abstracts (Jan 2017 by Ramin Homayouni, Diem-Trang Pham, and Sujoy Roy). For example, about 2500 PubMed abstracts contain reference to the gene "Sonic hedgehog" (Shh) in mouse, human, or rat. The words in all of these abstracts were extracted and categorize by their information content. A word such as "the" is not interesting, but words such as "dopamine" or "development" are useful in quantifying similarity. Sets of informative words are then compared???one gene's word set is compared the word set for all other genes. Similarity values are computed for a matrix of about 20,000 genes using latent semantic indexing [(see Xu et al., 2011)](http://journals.plos.org/plosone/article?id=10.1371/journal.pone.0018851). Similarity values are also known as literature correlations. These values are always positive and range from 0 to 1. Values between 0.5 and 1.0 indicate moderate-to-high levels of overlap of vocabularies. +The literature correlation is a unique feature in GeneNetwork that quantifies the similarity of words used to describe genes and their functions. Sets of words associated with genes were extracted from MEDLINE/PubMed abstracts (Jan 2017 by Ramin Homayouni, Diem-Trang Pham, and Sujoy Roy). For example, about 2500 PubMed abstracts contain reference to the gene "Sonic hedgehog" (Shh) in mouse, human, or rat. The words in all of these abstracts were extracted and categorize by their information content. A word such as "the" is not interesting, but words such as "dopamine" or "development" are useful in quantifying similarity. Sets of informative words are then compared--one gene's word set is compared the word set for all other genes. Similarity values are computed for a matrix of about 20,000 genes using latent semantic indexing [(see Xu et al., 2011)](http://journals.plos.org/plosone/article?id=10.1371/journal.pone.0018851). Similarity values are also known as literature correlations. These values are always positive and range from 0 to 1. Values between 0.5 and 1.0 indicate moderate-to-high levels of overlap of vocabularies. -The literature correlation can be used to compare the "semantic" signal-to-noise of different measurements of gene, mRNA, and protein expression. Consider this common situation:There are three probe sets that measure Kit gene expression (1459588\_at, 1415900\_a\_at, and 1452514\_a\_at) in the Mouse BXD Lung mRNA data set (HZI Lung M430v2 (Apr08) RMA). Which one of these three gives the best measurement of Kit expression? It is impractical to perform quantitative rtPCR studies to answer this question, but there is a solid statistical answer that relies on **Literature Correlation**. Do the following: For each of the three probe sets, generate the top 1000 literature correlates. This will generate three apparently identical lists of genes that are known from the PubMed literature to be associated with the Kit oncogene. But the three lists are NOT actually identical when we look at the **Sample Correlation** column. To answer the question "which of the three probe sets is best", review the actual performance of the probe sets against this set of 1000 "friends of Kit". Do this by sorting all three lists by their Sample Correlation column (high to low). The clear winner is probe set 1415900_a_at. The 100th row in this probe set's list has a Sample Correlation of 0.620 (absolute value). In comparison, the 100th row for probe set 1452514_a_at has a Sample Correlation of 0.289. The probe set that targets the intron comes in last at 0.275. In conclusion, the probe set that targets the proximal half of the 3' UTR (1415900_a_at) has the highest "agreement" between Literature Correlation and Sample Correlation, and is our preferred measurement of Kit expression in the lung in this data set. (Updated by RWW and Ramin Homayouni, April 2017.) +The literature correlation can be used to compare the "semantic" signal-to-noise of different measurements of gene, mRNA, and protein expression. Consider this common situation:There are three probe sets that measure Kit gene expression (1459588\_at, 1415900\_a\_at, and 1452514\_a\_at) in the Mouse BXD Lung mRNA data set (HZI Lung M430v2 (Apr08) RMA). Which one of these three gives the best measurement of Kit expression? It is impractical to perform quantitative rtPCR studies to answer this question, but there is a solid statistical answer that relies on **Literature Correlation**. Do the following: For each of the three probe sets, generate the top 1000 literature correlates. This will generate three apparently identical lists of genes that are known from the PubMed literature to be associated with the Kit oncogene. But the three lists are NOT actually identical when we look at the **Sample Correlation** column. To answer the question "which of the three probe sets is best", review the actual performance of the probe sets against this set of 1000 "friends of Kit". Do this by sorting all three lists by their Sample Correlation column (high to low). The clear winner is probe set 1415900_a_at. The 100th row in this probe set's list has a Sample Correlation of 0.620 (absolute value). In comparison, the 100th row for probe set 1452514_a_at has a Sample Correlation of 0.289. The probe set that targets the intron comes in last at 0.275. In conclusion, the probe set that targets the proximal half of the 3' UTR (1415900_a_at) has the highest "agreement" between Literature Correlation and Sample Correlation, and is our preferred measurement of Kit expression in the lung in this data set. (Updated by RWW and Ramin Homayouni, April 2017.) <div id="LOD"></div> @@ -327,16 +327,16 @@ A LOD score is defined as the logarithm of the ratio of two likelihoods: (1) in { pchisq(x*(2*log(10)),df=1,lower.tail=FALSE)/2 } - # (from https://www.biostars.org/p/88495/ ) + # (from https://www.biostars.org/p/88495/ ) </pre> In the two likelihoods, one has maximized over the various nuisance parameters (the mean phenotypes for each genotype group, or overall for the null hypothesis, and the residual variance). Or one can say, one has plugged in the maximum likelihood estimates for these nuisance parameters. With complete genotype data for a marker, the log likelihood for the normal model reduces to (-n/2) times the log of the residual sum of squares. -LOD values can be converted to LRS scores (likelihood ratio statistics) by multiplying by 4.61. The LOD is also roughly equivalent to the -log(P) when the degrees of freedom of the mapping has two degrees of freedom, as in a standard F2 intercross. In such as case, where P is the probability of linkage (P = 0.001) the ???logP => 3), will also equal a LOD of 3. The LOD itself is not a precise measurement of the probability of linkage, but in general for F2 crosses and RI strains, values above 3.3 will usually be worth attention for simple interval maps. +LOD values can be converted to LRS scores (likelihood ratio statistics) by multiplying by 4.61. The LOD is also roughly equivalent to the -log(P) when the degrees of freedom of the mapping has two degrees of freedom, as in a standard F2 intercross. In such as case, where P is the probability of linkage (P = 0.001) the -logP => 3), will also equal a LOD of 3. The LOD itself is not a precise measurement of the probability of linkage, but in general for F2 crosses and RI strains, values above 3.3 will usually be worth attention for simple interval maps. -LOD scores and ???logP scores are only interchangable when models have two degrees of freedom (2 df). +LOD scores and -logP scores are only interchangable when models have two degrees of freedom (2 df). Let us begin with an example. Suppose we have a LOD score of 3 from an F2 cross; this test has 2 df. Let us calculate the p-value (and the logp) corresponding to this LOD score. @@ -355,7 +355,7 @@ using Distributions # LOD score LOD = 3.0 -# p-value from chi-square with 2df calculated from +# p-value from chi-square with 2df calculated from # cdf (cumulative distribution function) p = 1-cdf(Chisq(2),2*log(10)*LOD) Out[1]: 0.0010000000000000009 @@ -396,7 +396,7 @@ The relationship between differences in a trait and differences in alleles at a </pre> To compute the marker regression (or correlation) we just compare values in Rows 2 and 4. A Pearson product moment correlation gives a value of r = 0.494. A regression analysis indicates that on average those strains with a D allele have a heavier brain with roughly a 14 mg increase for each 1 unit change in genotype; that is a total of about 28 mg if all B-type strains are compared to all D-type strains at this particular marker. This difference is associated with a p value of 0.0268 (two-tailed test) and an LRS of about 9.8 (LOD = 9.8/4.6 or about 2.1). Note that the number of strains is modest and the results are therefore not robust. If you were to add the two parent strains (C57BL/6J and DBA/2J) back into this analysis, which is perfectly fair, then the significance of this maker is lost (r = 0.206 and p = 0.3569). Bootstrap and permutation analyses can help you decide whether results are robust or not and whether a nominally significant p value for a single marker is actually significant when you test many hundreds of markers across the whole genome (a so-called genome-wide test with a genome-wide p value that is estimated by permutation testing). [RWW, Feb 20, 2007, Dec 14, 2012] - + [Go back to index](#index) <div id="n"></div> @@ -409,7 +409,7 @@ A [normal probability plot](http://en.wikipedia.org/wiki/Normal_probability_plot In genetic studies, the probability plot can be used to detect the effects of major effect loci. A classical Mendelian locus will typically be associated with either a bimodal or trimodal distribution. In the plot below based on 99 samples, the points definitely do not fall on a single line. Three samples (green squares) have unusually high values; the majority of samples fall on a straight line between z = -0.8 to z = 2; and 16 values have much lower trait values than would be predicted based on a single normal distribution (a low mode group). The abrupt discontinuity in the distribution at -0.8 z is due to the effect of a single major Mendelian effect. -Deviations from normality of the sort in the figure below should be considered good news from the point of view of likely success of tracking down the locations of QTLs. However, small numbers of outliers may require special statistical handling, such as their exclusion or [winsorising](http://en.wikipedia.org/wiki/Winsorising) (see more below on "Winsorizing"). [RWW June 2011] +Deviations from normality of the sort in the figure below should be considered good news from the point of view of likely success of tracking down the locations of QTLs. However, small numbers of outliers may require special statistical handling, such as their exclusion or [winsorising](http://en.wikipedia.org/wiki/Winsorising) (see more below on "Winsorizing"). [RWW June 2011] <img width="600px" src="/static/images/Normal_Plot.gif" alt="Homozygosity" /> @@ -477,12 +477,12 @@ An analysis of statistical power is useful to estimate numbers of replicates and #### [Power Calculator (D. Ashbrook)](https://dashbrook1.shinyapps.io/bxd_power_calculator_app/) -We can see that in all situations power is increased more by increasing the number of lines than by increasing the number of biological replicates. Dependent upon the heritability of the trait, there is little gain in power when going above 4-6 biological replicates. [DGA, Mar 3, 2018] +We can see that in all situations power is increased more by increasing the number of lines than by increasing the number of biological replicates. Dependent upon the heritability of the trait, there is little gain in power when going above 4-6 biological replicates. [DGA, Mar 3, 2018] #### Probes and Probe Sets: In microarray experiments the probe is the immobilized sequence on the array that is complementary to the target message washed over the array surface. Affymetrix probes are 25-mer DNA sequences synthesized on a quartz substrate. There are a few million of these 25-mers in each 120-square micron cell of the array. The abundance of a single transcript is usualy estimated by as many as 16 perfect match probes and 16 mismatch probes. The collection of probes that targets a particular message is called a probe set. [RWW, Dec 21, 2004] - + [Go back to index](#index) @@ -514,7 +514,7 @@ While the potential utility of RI strains in mapping complex polygenic traits wa By 2010 the number of mouse RI strains had increased to the point where defining causal gene and sequence variant was more practical. As of 2018 there are about 150 BXD strains (152 have been fully sequenced), ~100 Collaborative Cross strains (also all fully sequenced), and at least another 100 RI strains belonging to smaller sets that have been extremely well genotyped. -**Making RI strains**: The usual procedure typically involves sib mating of the progeny of an F1 intercross for more than 20 generations. Even by the 5th filial (F) generation of successive matings, the RI lines are homozygous at 50% of loci and by F13, the value is above 90%. At F20 the lines are nearly fully inbred (~98%) and by convention are now referred to as inbred strains rather than inbred lines. +**Making RI strains**: The usual procedure typically involves sib mating of the progeny of an F1 intercross for more than 20 generations. Even by the 5th filial (F) generation of successive matings, the RI lines are homozygous at 50% of loci and by F13, the value is above 90%. At F20 the lines are nearly fully inbred (~98%) and by convention are now referred to as inbred strains rather than inbred lines. <img width="600px" src="/static/images/SilverFig3_2.png" alt="" /> [Go back to index](#index) @@ -545,7 +545,7 @@ SNP is an acronym for single nucleotide polymorphisms (SNPs). SNPs are simple on The SNP track in WebQTL exploits the complete Celera Discovery System SNP set but adds an additional 500,000 inferred SNPs in both BXD and AXB/BXA crosses. These SNPs were inferred based on common haplotype structure using an Monte Carlo Markov chain algorithm developed by Gary Churchill and Natalie Blades and implemented by Robert Crowell, and RWW in July 2004. Raw data used to generate the SNP seismograph track were generated by Alex Williams and Chris Vincent, July 2003. The BXD track exploits a database of 1.75 million B vs D SNPs, whereas the AXB/BXA track exploits a database of 1.80 million A vs B SNPs. The names, sequences, and precise locations of most of these SNPs are the property of Celera Discovery Systems, whom we thank for allowing us to provide this level of display in WebQTL. -Approximately 2.8 million additional SNPs generated by Perlegen for the NIEHS have been added to the SNP track by Robert Crowell (July-Aug 2005). We have also added all Wellcome-CTC SNPs and all relevant mouse SNPs from dbSNP. [Williams RW, Dec 25, 2004; Sept 3, 2005] +Approximately 2.8 million additional SNPs generated by Perlegen for the NIEHS have been added to the SNP track by Robert Crowell (July-Aug 2005). We have also added all Wellcome-CTC SNPs and all relevant mouse SNPs from dbSNP. [Williams RW, Dec 25, 2004; Sept 3, 2005] #### Standard Error of the Mean (SE or SEM): @@ -555,11 +555,11 @@ where n is the number of independent biological samples used to estimate the pop #### Strain Distribution Pattern: -A marker such as a SNP or microsatellite is genotyped using DNA obtained from each member of the mapping population. In the case of a genetic reference population, such as the BXD strains or the BayXSha Arabadopsis lines, this results in a text string of genotypes (e.g., BDDDBDBBBBDDBDDDBBBB... for BXD1 through BXD100). Each marker is associated with its own particular text string of genotypes that is often called the **strain distribution pattern** of the marker. (A more appropriate term would be the **marker genotype string**.) This string is converted to a numerical version, a genotype vector: -1111-11-1-1-1-111-1111-1-1-1-1..., where D=1, B=-1, H=0. Mapping a trait boils down to performing correlations between each trait and all of the genotype vectors. The genotype vector with the highest correlation (absolute value) is a good candidate for a QTL. [Williams RW, June 18, 2005] +A marker such as a SNP or microsatellite is genotyped using DNA obtained from each member of the mapping population. In the case of a genetic reference population, such as the BXD strains or the BayXSha Arabadopsis lines, this results in a text string of genotypes (e.g., BDDDBDBBBBDDBDDDBBBB... for BXD1 through BXD100). Each marker is associated with its own particular text string of genotypes that is often called the **strain distribution pattern** of the marker. (A more appropriate term would be the **marker genotype string**.) This string is converted to a numerical version, a genotype vector: -1111-11-1-1-1-111-1111-1-1-1-1..., where D=1, B=-1, H=0. Mapping a trait boils down to performing correlations between each trait and all of the genotype vectors. The genotype vector with the highest correlation (absolute value) is a good candidate for a QTL. [Williams RW, June 18, 2005] #### Suggestive Threshold: -The suggestive threshold represents the approximate LRS value that corresponds to a genome-wide p-value of 0.63, or a 63% probability of falsely rejecting the null hypothesis that there is no linkage anywhere in the genome. This is not a typographical error. The Suggestive LRS threshold is defined as that which yields, on average, one false positive per genome scan. That is, roughly one-third of scans at this threshold will yield no false positive, one-third will yield one false positive, and one-third will yield two or more false positives. This is a very permissive threshold, but it is useful because it calls attention to loci that may be worth follow-up. Regions of the genome in which the LRS exceeds the suggestive threshold are often worth tracking and screening. They are particularly useful in combined multicross metaanalysis of traits. If two crosses pick up the same suggestive locus, then that locus may be significant when the joint probability is computed. The suggestive threshold may vary slightly each time it is recomputed due to the random generation of permutations. You can view the actual histogram of the permutation results by selecting the "Marker Regression" function in the **Analysis Tools** area of the **Trait Data and Editing Form**. [Williams RW and Manly KF, Nov 15, 2004] +The suggestive threshold represents the approximate LRS value that corresponds to a genome-wide p-value of 0.63, or a 63% probability of falsely rejecting the null hypothesis that there is no linkage anywhere in the genome. This is not a typographical error. The Suggestive LRS threshold is defined as that which yields, on average, one false positive per genome scan. That is, roughly one-third of scans at this threshold will yield no false positive, one-third will yield one false positive, and one-third will yield two or more false positives. This is a very permissive threshold, but it is useful because it calls attention to loci that may be worth follow-up. Regions of the genome in which the LRS exceeds the suggestive threshold are often worth tracking and screening. They are particularly useful in combined multicross metaanalysis of traits. If two crosses pick up the same suggestive locus, then that locus may be significant when the joint probability is computed. The suggestive threshold may vary slightly each time it is recomputed due to the random generation of permutations. You can view the actual histogram of the permutation results by selecting the "Marker Regression" function in the **Analysis Tools** area of the **Trait Data and Editing Form**. [Williams RW and Manly KF, Nov 15, 2004] #### Systems Genetics: @@ -583,7 +583,7 @@ The term "systems genetics" was coined by Grant Morahan, October 2004, during a The tissue correlation is an estimate of the similarity of expression of two genes across different cells, tissues, or organs. In order to compute this type of correlation we first generate expression data for multiple different cell types, tissues, or whole organs from a single individual. There will be significant differences in gene expression across this sample and this variation can then be used to compute either Pearson product-moment correlations (r) or Spearman rank order correlations (rho) between any pair of genes, transcripts, or even exons. Since the samples are ideally all from one individual there should not be any genetic or environmental differences among samples. The difficulty in computing tissue correlations is that samples are not independent. For example, three samples of the small intestine (jejunum, ilieum, and duodenum) will have expression patterns that are quite similar to each other in comparison to three other samples, such as heart, brain, and bone. For this reason the nature of the sampling and how those samples are combined will greatly affect the correlation values. The tissue correlations in GeneNetwork were computed in a way that attempts to reduce the impact of this fact by combining closely related sample types. For example multiple data sets for different brain region were combined to generate a single average CNS tissue sample (generating a whole brain sample would have been an alternative method). -However, there is really not optimal way to minimize the effects of this type of non-independence of samples. Some genes will have high expression in only a few tissues, for example the cholinergic receptor, nicotinic, alpha polypeptide 1 gene Chrna1 has high expression in muscle tissues (skeletal muscle = Mus, tongue = Ton, and esophagus = Eso) but lower expression in most other tissues. The very high correlation between Chrna1 and other genes with high expression only in muscle reflects their joint bimodality of expression. It does not mean that these genes or their proteins necessarily cooperate directly in molecular processes. [Williams RW, Dec 26, 2008] +However, there is really not optimal way to minimize the effects of this type of non-independence of samples. Some genes will have high expression in only a few tissues, for example the cholinergic receptor, nicotinic, alpha polypeptide 1 gene Chrna1 has high expression in muscle tissues (skeletal muscle = Mus, tongue = Ton, and esophagus = Eso) but lower expression in most other tissues. The very high correlation between Chrna1 and other genes with high expression only in muscle reflects their joint bimodality of expression. It does not mean that these genes or their proteins necessarily cooperate directly in molecular processes. [Williams RW, Dec 26, 2008] <img width="600px" src="/static/images/Chrna1vsMyf6.gif" alt="" /> @@ -593,14 +593,14 @@ The small orange triangle on the x-axis indicates the approximate position of th #### Transform: -Most of the data sets in the GeneNetwork are ultimately derived from high resolution images of the surfaces of microarrays. Estimates the gene expression therefore involves extensive low-level image analysis. These processesing steps attempt to compensate for low spatial frequency "background" variation in image intensity that cannot be related to the actual hybridization signal, for example, a gradation of intensity across the whole array surface due to illumination differences, uneven hybridization, optical performance, scanning characteristics, etc. High spatial frequeny artifacts are also removed if they are likely to be artifacts: dust, scrathes on the array surface, and other "sharp" blemishes. The raw image data (for example, the Affymetrix DAT file) also needs to be registered to a template that assigns pixel values to expected array spots (cells). This image registration is an important process that users can usually take for granted. The end result is the reliable assignment of a set of image intensity values (pixels) to each probe. Each cell value generated using the Affymetrix U74Av2 array is associated with approximately 36 pixel intensity values (a 6x6 set of pixels, usually an effective 11 or 12-bit range of intensity). Affymetrix uses a method that simply ranks the values of these pixels and picks as the "representative value" the pixel that is closest to a particular rank order value, for example, the 24th highest of 36 pixels. The range of variation in intensity values amoung these ranked pixels provides a way to estimate the error of the estimate. The Affymetrix CEL files therefore consist of XY coordinates, the consensus value, and an error term. [Williams RW, April 30, 2005] +Most of the data sets in the GeneNetwork are ultimately derived from high resolution images of the surfaces of microarrays. Estimates the gene expression therefore involves extensive low-level image analysis. These processesing steps attempt to compensate for low spatial frequency "background" variation in image intensity that cannot be related to the actual hybridization signal, for example, a gradation of intensity across the whole array surface due to illumination differences, uneven hybridization, optical performance, scanning characteristics, etc. High spatial frequeny artifacts are also removed if they are likely to be artifacts: dust, scrathes on the array surface, and other "sharp" blemishes. The raw image data (for example, the Affymetrix DAT file) also needs to be registered to a template that assigns pixel values to expected array spots (cells). This image registration is an important process that users can usually take for granted. The end result is the reliable assignment of a set of image intensity values (pixels) to each probe. Each cell value generated using the Affymetrix U74Av2 array is associated with approximately 36 pixel intensity values (a 6x6 set of pixels, usually an effective 11 or 12-bit range of intensity). Affymetrix uses a method that simply ranks the values of these pixels and picks as the "representative value" the pixel that is closest to a particular rank order value, for example, the 24th highest of 36 pixels. The range of variation in intensity values amoung these ranked pixels provides a way to estimate the error of the estimate. The Affymetrix CEL files therefore consist of XY coordinates, the consensus value, and an error term. [Williams RW, April 30, 2005] #### Transgression: Most of us are familiar with the phrase "regression toward the mean." This refers to the tendency of progeny of a cross to have phenotype that are intermediate to those of the parents. Transgression refers to the converse: progeny that have more phenotypes that are higher and lower than those of either parent. Transgression is common, and provided that a trait is influenced by many independent sequence variants (a polygenic trait), transgression is the expectation. This is particularly true if the parents are different genetically, but by chance have similar phenotypes. Consider a trait that is controlled by six independent genes, A through F. The "0" allele at these size genes lowers body weight whereas the "1" allele increases body weight. If one parent has a 000111 6-locus genotype and the other parent has 111000 genotype, then they will have closely matched weight. But their progeny may inherit combinations as extreme as 000000 and 111111. Transgression means that you can rarely predict the distribution of phenotypes among a set of progeny unless you already have a significant amount of information about the genetic architecture of a trait (numbers of segregating variants that affect the trait, either interactions, and GXE effects). In practical terms this means that if the parents of a cross do NOT differ and you have good reasons to believe that the trait you are interested in is genetically complex, then you can be fairly confident that the progeny will display a much wider range of variation that the parents. [May 2011 by RWW]. - + [Go back to index](#index) <div id="u"></div> diff --git a/general/help/facilities.md b/general/help/facilities.md index 7234059..7074a7d 100644 --- a/general/help/facilities.md +++ b/general/help/facilities.md @@ -1,63 +1,37 @@ # Equipment -The core [GeneNetwork team](https://github.com/genenetwork/) and [Pangenome -team](https://github.com/pangenome) at UTHSC maintains modern Linux servers -and storage systems for genetic, genomic, pangenome, pangenetics, and -phenome analyses. -Machines are located in in the main UTHSC machine room of the Lamar -Alexander Building at UTHSC (Memphis TN campus). This is a physically -secure location with raised -floors and an advanced fire extinguishing system. - +The core [GeneNetwork team](https://github.com/genenetwork/) and [Pangenome team](https://github.com/pangenome) at UTHSC maintains modern Linux servers and storage systems for genetic, genomic, pangenome, pangenetics, and phenome analyses. Machines are located in in the main UTHSC machine room of the Lamar Alexander Building at UTHSC (Memphis TN campus). +This is a physically secure location with raised floors and an advanced fire extinguishing system. We have access to this space for upgrades and hardware maintenance. -We use remote racadm and/or ipmi to all machines for out-of-band -maintenance. -Issues and work packages are tracked through our 'tissue' [tracker board]( -https://issues.genenetwork.org/) and we use git repositories for -documentation, issue tracking and planning (mostly public and some private -repos available on request). -We also run [continuous integration](https://ci.genenetwork.org/) and -[continuous deployment](https://cd.genenetwork.org/) services online (CI -and CD). At FOSDEM 2023 -Arun Isaac presented tissue, our [minimalist git+plain text issue tracker](https://archive.fosdem.org/2023/schedule/event/tissue/) that allows us to -move away from github soure code hosting and issue trackers. - -The computing facility has four computer racks dedicated to -GeneNetwork-related work and pangenomics. -Each rack has a mix of Dell PowerEdge servers (from a few older low-end -R610s, R6515, and two R7425 AMD Epyc 64-core 256GB RAM systems - tux01 and -tux02 - running the GeneNetwork web services). -We also support several experimental systems, including a 40-core R7425 -system with 196 GB RAM and 2x NVIDIA V100 GPU (tux03), and one Penguin -Computing Relion 2600GT systems (Penguin2) with NVIDIA Tesla K80 GPU used -for software development and to serve outside-facing less secure R/shiny -and Python services that run in isolated containers. Effectively, we have -three outward facing servers that are fully utilized by the GeneNetwork -team with a total of 64+64+40+28 = 196 real cores. -In 2023 we added two machines to upgrade from tux01 and tux02 -- named -tux04 and tux05 resp. --- that have the latest Dell Poweredge R6625 AMD -Genoa EPYC processors adding a total of 96 real CPU cores running at 4GHz. -These two machines have 768Gb RAM each. + +We use remote racadm and/or ipmi to all machines for out-of-band maintenance. +Issues and work packages are tracked through our 'tissue' [tracker board](https://issues.genenetwork.org/) and we use git repositories for documentation, issue tracking and planning (mostly public and some private repos available on request). +We also run [continuous integration](https://ci.genenetwork.org/) and [continuous deployment](https://cd.genenetwork.org/) services online (CI and CD). +At FOSDEM 2023 Arun Isaac presented tissue, our [minimalist git+plain text issue tracker](https://archive.fosdem.org/2023/schedule/event/tissue/) that allowed us to move away from github soure code hosting and github issue trackers. + +The computing facility has four computer racks dedicated to GeneNetwork-related work and pangenomics. Each rack has a mix of Dell PowerEdge servers (from a few older low-end R610s, R6515, and three R7425 AMD Epyc systems - tux01 and tux02 - running development and experimental web services). In 2023 we added two machines to upgrade from tux01 and tux02 -- named tux04 and tux05 resp. --- that have the latest Dell Poweredge R6625 AMD Genoa EPYC processors adding a total of 96 real CPU cores running at 4GHz. These two machines have 768Gb RAM each. +We also support several experimental systems, including a 40-core R7425 system with 196 GB RAM and 2x NVIDIA V100 GPU (tux03), and one Penguin Computing Relion 2600GT systems (Penguin2) with NVIDIA Tesla K80 GPU used for software development and to serve outside-facing less secure R/shiny and Python services that run in isolated containers. +Effectively, we have outward facing servers that are fully utilized by the GeneNetwork team with a total of 64+64+40+28 = 196 real cores. ## Octopus HPC cluster -In 2020 we installed a powerful HPC cluster (Octopus) dedicated to -[pangenomic](https://www.biorxiv.org/content/10.1101/2021.11.10.467921v1) +In 2020 we installed a small but powerful HPC cluster (Octopus) dedicated to our +[pangenomic](https://pubmed.ncbi.nlm.nih.gov/39433878/) and [genetic](https://genenetwork.org/) computations, consisting of 11 PowerEdge R6515 AMD EPYC 7402P 24-core CPUs (264 real cores). -In 2023 we added 4 new R6625 AMD Genoa machines adding a total of 192 real -CPU cores running at 4GHz (total of 438 real CPU cores). -Nine of these machines are equipped with 378 GB RAM, four R6625 have 768 GB -and two have 1 TB of memory. -All machines have large SSD storage (~10TB) driving the MooseFS shared -network storage. MooseFS is configured with three storage classes: 2CP +In 2023 we added 5 new R6625 AMD Genoa machines adding a total of 240 real +CPU cores running at 4GHz (i.e., a total of 504 real CPU cores). +Nine of these machines are equipped with 378 GB RAM, five R6625 have 768 GB +and two R6515 have 1 TB of memory. +All machines have large SSD storage (~10TB) driving the MooseFS distributed shared +network storage. MooseFS is flexible in its storage policies. MooseFS is configured with three storage classes: 2CP (default) with one copy on SSD and one on RAID5 spinning HDD for redundancy, scratch for fast non-redundant SSD access, and raid5 for -archival storage on spinning disks. +archival storage on spinning disks. For high throughput we also define multiple copies on the network storage (e.g. 4CP). All Octopus nodes run Debian and GNU Guix and use Slurm for batch submission. -We run MooseFS for distributed network file storage and we run the common -workflow language (CWL), Docker, and Apptainer containers. +We can run the common +workflow language (CWL), Docker, and Apptainer containers amongst other solutions. The racks have dedicated 10Gbs high-speed Cisco switches and firewalls that are maintained by UTHSC IT staff. This heavily used cluster, notably, is almost self-managed by its users and @@ -69,8 +43,8 @@ This heavily used cluster, notably, is almost self-managed by its users and activity reports! The total number of cores for Octopus has essentially doubled to a total of -456 real CPU cores and the MooseFS SSD distributed network storage is -getting close to 200TB with fiber optic interconnect. +504 real CPU cores and the MooseFS SSD distributed network storage is +getting close to 250TB with fiber optic interconnect. <table border="0" style="width:95%"> <tr> @@ -118,10 +92,8 @@ computing facility operated by the UT Joint Institute for Computational Sciences in a secure setup at the DOE Oak Ridge National Laboratory (ORNL) and on the UT Knoxville campus. We have a 10 Gbit connection from the machine room at UTHSC to data -transfer nodes at ISAAC. ISAAC is continuously being upgraded (see [ISAAC -system overview](https://oit.utk.edu/hpsc/available-resources/)) and has -over 7 PB of high-performance Lustre DDN storage and contains over 20,000 -cores with some large RAM nodes and 32 GPU nodes. +transfer nodes at ISAAC. ISAAC is continuously being upgraded (see [ISAAC system overview](https://oit.utk.edu/hpsc/available-resources/)) and has +over 7 PB of high-performance Lustre DDN storage and contains over 25,000 cores with some large RAM nodes and 33 GPU nodes. Drs. Prins, Garrison, Colonna, Chen, Ashbrook and other team members use ISAAC systems to analyze genomic and genetic data sets. Note that we can not use ISAAC and storage facilities for public-facing web |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}